

ISO 13485 is the internationally recognised standard for quality management systems (QMS) in the medical device sector. It sets out what manufacturers must do to ensure their devices consistently meet regulatory requirements and customer expectations. The standard provides the structure for building a system that fosters control, traceability, and ongoing improvement across all levels of a business.
This article explains the core requirements of ISO 13485, including documentation, responsibilities, procedural controls, and how the standard connects to wider regulatory obligations.
The core requirements of ISO 13485 include:
Some requirements of ISO 13485, such as those relating to SOPs, are only briefly outlined in this article, in the interest of focus and clarity.
Every certified QMS begins with a quality manual. It defines how the company approaches quality, describes the scope of the QMS, and sets out quality policy and objectives.
The manual should include:
Although it’s possible to embed SOPs directly into the quality manual, most companies opt to keep these documents separate for ease of maintenance and clarity during audits.
SOPs form the procedural core of ISO 13485. These documents define how key activities must be performed to ensure consistency and compliance. They are required for areas such as:

SOPs must be trained, followed, and periodically reviewed for effectiveness. Employees should be able to demonstrate knowledge of the procedures relevant to their roles.
One of the defining features of ISO 13485 is the integration of risk management into all QMS processes, not just product development. This is not optional. Risk-based thinking must underpin decisions around:
Clause 4.1.2 requires manufacturers to apply a risk-based approach to control the appropriate processes needed for the QMS. This expectation ties directly into ISO 14971 for product risk, but also applies to quality and organisational risks.
Regular internal audits (Clause 8.2.4) are an essential part of ISO 13485 and a basic requirement for QMS certification. These audits are structured reviews, conducted by trained auditors, to assess whether processes are being implemented as intended. As the name suggests, internal audits are conducted within the organisation, in contrast to external audits, which are carried out to demonstrate formal compliance. Internal audits help prepare for those external assessments, but they also play a critical role in identifying weaknesses early, preventing errors, and improving processes in a focused and systematic way.
Audits are conducted according to a defined audit programme that ensures all relevant areas of the QMS are reviewed within a given timeframe. The frequency depends on both risk and significance: processes that have a greater impact on safety or regulatory compliance require more frequent review. Each audit must be thoroughly planned and documented, including the audit method, evaluation criteria, individuals involved, and outcomes. Any nonconformities identified feed directly into the CAPA process and serve as a key input for management review.
To ensure meaningful results, audits must be carried out by qualified and independent personnel who do not evaluate their own work and who avoid a purely procedural approach. Where necessary, internal audits may also be conducted by external professionals, provided they understand the organisation’s context and operate without conflict of interest.
The QMS must include formalised processes for gathering and analysing feedback and complaints (Clause 8.2.1–8.2.3). This is essential for identifying both actual and potential nonconformities.
Post-market surveillance data, such as clinical feedback or adverse event reports, must feed into:
Under the MDR, the QMS must also support Post-Market Clinical Follow-Up (PMCF) and Periodic Safety Update Reports (PSURs), activities that go beyond ISO 13485 alone but rely on its structure.
According to ISO 13485, every organization must appoint a quality management representative (QMR) (Clause 5.5.2). This person, positioned at management level, is responsible for ensuring that the quality management system (QMS) is properly implemented. Their responsibilities include meeting regulatory requirements and making sure that employees understand quality management and their specific roles within it.
The QMR must be part of the management team, but does not need to belong to top management. However, they must have sufficient authority to act independently and without any conflicts of interest. It is not necessary for quality management to be their sole area of responsibility, they may hold additional roles within the organisation. While day-to-day tasks can be delegated, the QMR remains ultimately accountable for the integrity and performance of the QMS. They must have excellent knowledge of the QMS and a deep understanding of the company’s products. Strong communication and leadership abilities are also essential for the role.
ISO 13485 allows certain clauses to be excluded, but only if they clearly do not apply to the company’s activities. For example:
In both cases, these decisions must be documented with a clear justification in the quality manual. Auditors will expect to see this reasoning during reviews.
ISO 13485 is designed to be compatible with other regulatory systems. It requires companies to identify and integrate relevant legal and technical standards into their QMS. Examples include:
These external requirements must be actively integrated into the QMS. Simply complying with ISO 13485 is not enough if additional obligations from the device’s target market are not addressed.
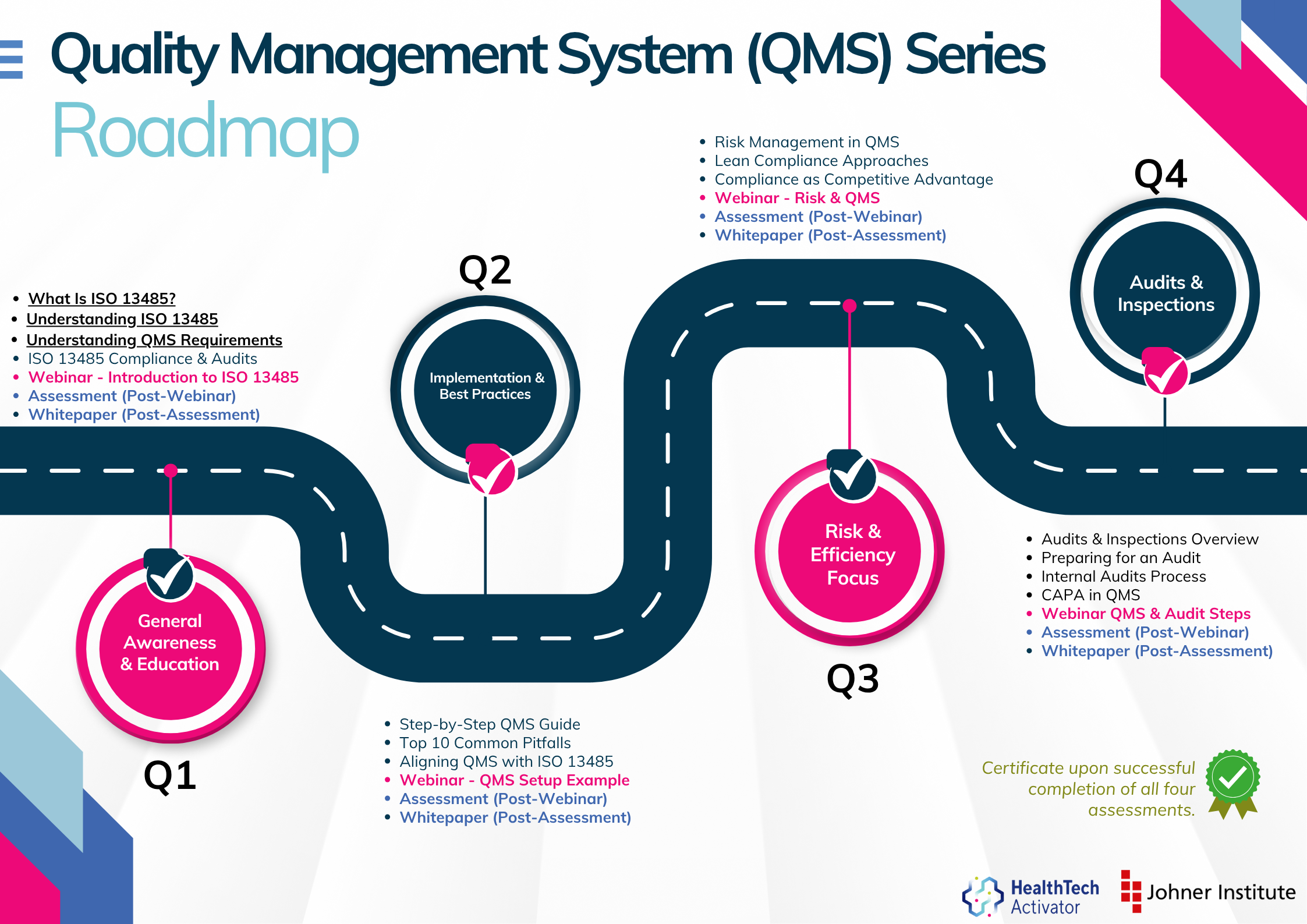
Meeting the requirements of ISO 13485 is one thing, putting them into practice is another. That’s why this article is part of a broader series developed for New Zealand’s HealthTech sector, aimed at helping teams turn regulatory expectations into working systems.
The QMS Series is brought to you by the HealthTech Activator, in partnership with the Johner Institute New Zealand.
This series explores ISO 13485 in four parts: from first steps and system setup to risk management and audit readiness. Participants who complete the quiz at the end of each section will receive an ISO 13485 QMS Certificate of Completion from the HealthTech Activator.

