
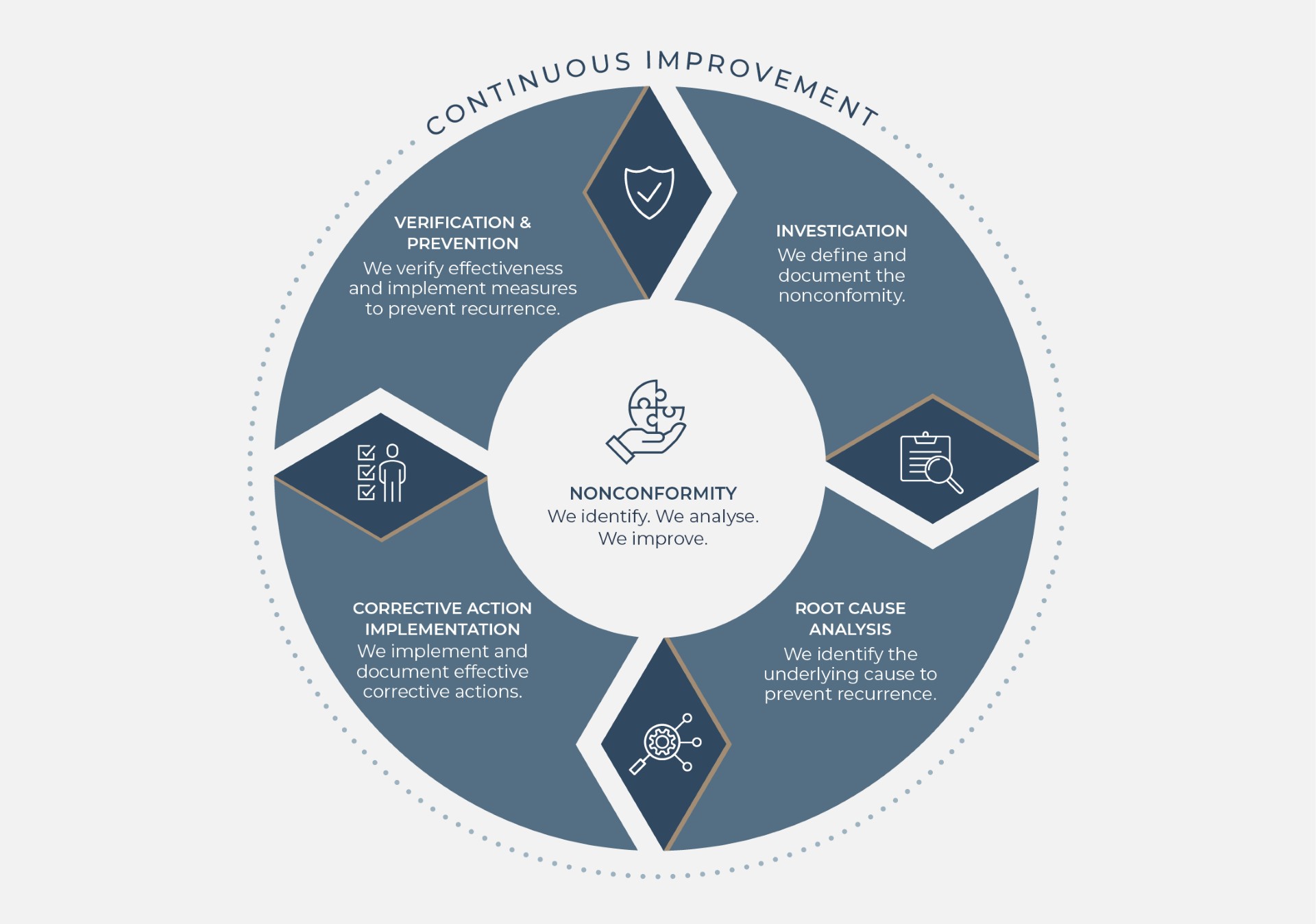
Corrective and preventive action (CAPA) is one of the mechanisms that turns a quality management system from a set of documents into a system that learns. In an ISO 13485-compliant QMS, CAPA helps organisations address nonconformities, investigate why they happened, implement actions that are proportionate to risk, and check whether those actions have actually worked.
For small and growing medical device companies, CAPA is especially important because recurring issues consume time, create avoidable cost, and can undermine confidence in the QMS. A well-designed CAPA process supports continuous improvement by linking complaints, nonconformities, audit findings, supplier issues, trend data, and post-market information to practical action before problems become systemic.
A common misunderstanding is to treat CAPA as a paperwork exercise that starts and ends with an isolated fix. In practice, ISO 13485 expects organisations to distinguish between immediate correction, corrective action that addresses the cause of a detected issue, and preventive action that reduces the likelihood of potential issues occurring elsewhere in the system.
This distinction matters because recurring issues rarely come from a single operator error or one defective batch alone. They often point to a deeper weakness in training, process design, supplier controls, design transfer, change management, or the way data is reviewed and escalated inside the QMS.
An effective CAPA process begins with clear inputs. These may include customer complaints, internal audit findings, product or process nonconformities, service reports, supplier performance issues, management review outputs, or trend signals identified through data analysis.
Once an issue is identified, the organisation should assess significance and risk, contain the problem where necessary, and decide whether formal CAPA is required. Not every minor event needs a full investigation, but repeated minor events, or a single event with high patient, user, or compliance impact, should trigger a structured response.
A practical CAPA workflow usually includes:
Recurring issues often occur when organisations address the symptom of a problem but not the underlying cause. For example, replacing a failed component may resolve the immediate product problem issue, but it will not prevent recurrence if the real cause is inadequate supplier qualification, unclear specifications, or weak incoming inspection criteria.
Another common contributor is poor problem definition at the start of the CAPA process. If teams launch action before understanding where, when, and how often the problem occurs, they may implement solutions that are easy to execute but ineffective in practice.
Weak effectiveness checks can also lead to repeated issues. Closing a CAPA immediately after training has been delivered or a form has been updated does not demonstrate that the problem has been resolved. Organisations need objective evidence that the action was effective, such as improved trends, reduced repeat deviations, or audit confirmation after implementation.
For start-ups and small manufacturers, the most useful CAPA systems are usually simple, risk-based, and well connected to other QMS processes. The process should be easy for staff to use, but structured enough to support escalation, investigation depth, and management visibility when issues suggest a broader system failure.
Several practices help:
When CAPA works well, it does more than close nonconformities. It creates feedback loops that help organisations learn from internal failures, field experience, and near misses, which is central to maintaining an effective and compliant quality management system over time.
In that sense, CAPA is not just a regulatory expectation. It is a practical discipline for avoiding repeat problems, protecting product quality, and building confidence that the QMS can support safe and scalable growth.
Meeting the requirements of ISO 13485 is one thing, putting them into practice is another. That’s why this article is part of a broader series developed for New Zealand’s HealthTech sector, aimed at helping teams turn regulatory expectations into working systems.
This series explores ISO 13485 in four parts: from first steps and system setup to risk management and audit readiness. Participants who complete the quiz at the end of each section will receive an ISO 13485 QMS Certificate of Completion from the HealthTech Activator.
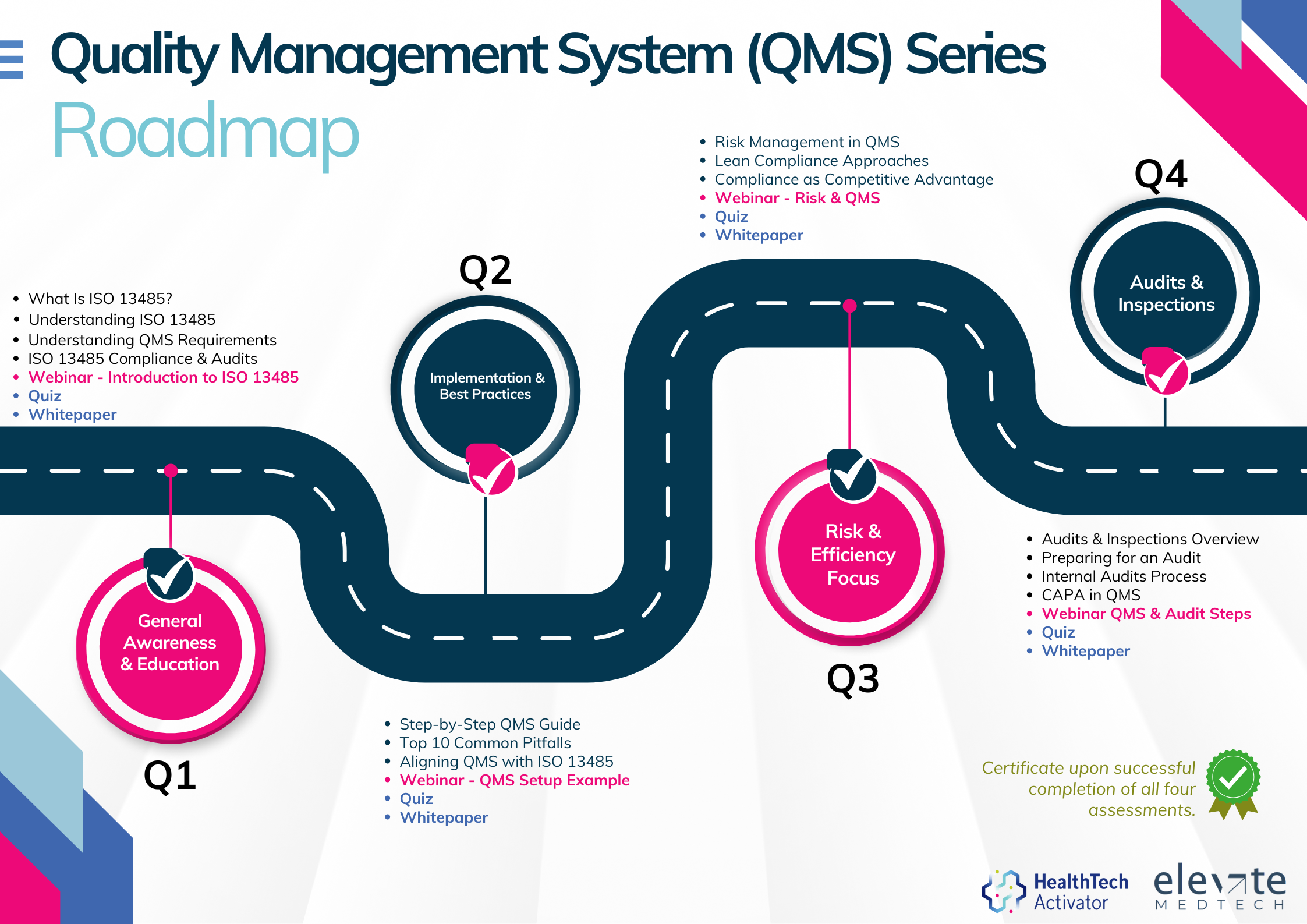
The QMS Series is brought to you by the HealthTech Activator, in partnership with Elevate Medtech.

