

A quality management system (QMS) according to ISO 13485 is legally required in almost every jurisdiction to bring a medical device to market. But treating it as merely a regulatory obligation means missing out on significant advantages. It is time to change perspective and fully exploit the benefits ISO 13485 offers beyond compliance. This article shows how to turn your QMS into a real competitive advantage.
The best marketing is a good product. Your ISO 13485 QMS is valuable in two ways. First, it improves overall product quality, which is one of the most important criteria for customers, partners, and patients when choosing and using a medical device. Certification and a mature QMS are often selection criteria for hospitals, original equipment manufacturer (OEM) partners, and distributors; being certified early can put you on shortlists competitors cannot reach. For startups, being audit-ready reassures investors and strategic partners that scaling up carries lower risk. ISO 13485 encourages clear definition and monitoring of processes, and metrics such as cycle time, first-pass yield, or CAPA closure time become concrete evidence of operational excellence.
Second, the certification itself is a strong external signal. High-quality products are not always obvious at first glance, but ISO 13485 certification visibly demonstrates adherence to strict quality standards. This builds trust and trust ensures that customers choose your products over competitors’, often for more than a single product. Once customers trust your company, they usually remain loyal. The takeaway: focus on delivering the highest product quality through your QMS and make that quality visible externally by highlighting your ISO 13485 certification.
A well-run QMS ensures processes run efficiently. This not only improves quality and safety but also removes redundancies, reducing costs and speeding up work. Documentation also helps when onboarding new employees, preventing mistakes and improving efficiency.
In product development, a risk-based QMS reduces rework and late-stage findings by highlighting critical issues earlier, when corrections are cheaper. Prioritising engineering, validation, and supplier controls for high-risk components, while applying lighter controls to low-risk areas, frees capacity and keeps development flowing smoothly.
ISO 13485’s documentation requirements provide another advantage. Complaints, feedback, and real-world performance data can be turned into structured input for product roadmaps and feature decisions. Companies that systematically mine post-market surveillance (PMS) data can respond faster to market needs and differentiate on reliability and usability. Treat PMS as a continuous product intelligence loop, not a passive compliance task. Standardise how data is collected, analysed, acted upon, and fed back into design, usability, and marketing.
The notion that strict quality management and innovation do not go together is outdated. On the contrary, innovation can be baked into the QMS. Quality by design and risk-based controls support faster, safer iteration rather than blocking it. A well-run QMS helps justify bolder designs to regulators because evidence, rationale, and traceability are already in place.
These are not just theoretical ideas. Different companies already use their QMS as a market advantage. Here is how:
Context
A cardiology-focused Software as a Medical Device (SaMD) manufacturer develops diagnostic and monitoring software used to support clinical decisions. Because specific software functions can influence diagnoses and treatment, the organisation applies a structured, risk-based approach to change control aligned with ISO 13485, ISO 14971 and IEC 62304.
Approach
Functionalities are classified into high-risk and low-risk categories based on their potential impact on patient safety and clinical performance. High-risk elements, such as diagnostic algorithms, clinical decision-support logic and alarm behaviour, are subject to full design control, including documented design inputs, verification and validation activities using appropriate clinical or representative datasets, and complete traceability from requirements to test evidence. Low-risk elements, such as visual presentation, navigation, non-clinical dashboards or administrative exports, follow a predefined, simplified pathway that maintains control while reducing administrative burden.
Process
Impact
This tiered model enables frequent, controlled updates to user interface and usability aspects without diluting the rigor applied to safety-critical functions. It supports agile development practices while maintaining robust evidence for regulated clinical claims and ensuring continued conformity with software lifecycle and risk-management standards.
Context
A startup developing a wearable device for continuous physiological monitoring seeks early acceptance by hospital groups and institutional buyers. Demonstrable control over quality and reliability is essential, but resources are limited, making a lean, risk-based Quality Management System (QMS) the practical choice.
Approach
The company implements a “right-sized” ISO 13485 QMS that concentrates controls where risk is highest—design and development, purchasing, production, complaint handling and post-market surveillance—while keeping low-risk processes deliberately lightweight. Documentation is standardised through concise templates, and process design emphasises clarity, traceability and responsiveness over volume of paperwork.
Process
Impact
During tenders with hospital groups, the company can provide objective evidence of short complaint-resolution times, structured and up-to-date risk management documentation, and robust supplier controls linked to device reliability and continuity of supply. This positions the QMS not merely as a compliance obligation but as a tangible risk-mitigation measure for the customer, strengthening the startup’s competitive posture in procurement processes.
Context
An early-stage in vitro diagnostic (IVD) manufacturer needs to implement post-market surveillance (PMS) in line with ISO 13485 and IVDR expectations, while staying within the constraints of a small team and a limited installed base.
Approach
The organisation adopts a deliberately simple but systematic PMS framework, centred on structured feedback collection, periodic multidisciplinary review and traceable follow-up actions. The design of the process reflects regulatory expectations that PMS should gather user experience, feed into risk management and drive iterative improvement of device performance and usability.
Process
Impact
This proportionate PMS system satisfies regulatory expectations for continuous post-market monitoring, trend analysis and feedback integration, while remaining manageable in a resource-constrained environment. At the same time, the company can credibly present interface and workflow refinements as evidence that later product versions are directly informed by real-world user feedback, thereby reducing support burden and strengthening market acceptance, market monitoring, trend analysis and feedback integration, while remaining manageable in a resource-constrained environment.
Context
A global original equipment manufacturer (OEM) with a broad supplier base and multiple manufacturing sites faced increasing complexity in its quality and purchasing controls. Traditional, uniform application of procedures to all suppliers created audit bottlenecks, extended qualification timelines and diverted resources away from truly critical products and components.
Approach
The organisation redesigned its supplier and process controls using a risk-based tiering model. Suppliers and associated processes were categorised according to their impact on product safety, regulatory compliance and business continuity. High-risk and strategically critical suppliers received intensified oversight, while low-risk, low-impact contract manufacturers were managed through simplified controls.
Process
Impact
The risk-based re-tiering reduced unnecessary audit and documentation effort for low-risk suppliers and removed recurring approval bottlenecks. Vendor qualification for routine suppliers became faster, while the organisation freed capacity to rigorously oversee critical suppliers and support high-risk new product introductions. Compliance activities thereby contributed directly to throughput and time-to-market, rather than acting as a constraint.
Context
An established medical device manufacturer with a mature QMS needed to increase the effectiveness of its internal audit programme. Traditional, schedule-driven audits generated findings but had limited impact on strategic priorities, such as managing emerging risks and demonstrating reliability improvements to key customers.
Approach
The company repositioned internal audits as a targeted risk and improvement tool rather than a purely cyclical compliance activity. Audit planning was driven by process risk, regulatory exposure and strategic importance, with particular emphasis on areas where failure could significantly affect patient safety, cybersecurity, or service quality for major accounts.
Process
Impact
The refocused internal audit programme generated findings that directly informed improvement projects in high-risk processes and customer-critical areas. Over time, this contributed to measurable gains in reliability and service performance. The organisation was able to present this structured audit and improvement cycle as evidence of ongoing risk management and reliability enhancement in communications with strategic customers and procurement stakeholders.
To establish your QMS as a market advantage, treat audits as short, focused improvement projects targeting your riskiest and most strategic processes:
ISO 13485 does not have to be a burden. When leveraged strategically, it improves product quality, operational efficiency, data-driven decision-making, and innovation. Companies that embrace ISO 13485 convert compliance into a tangible market advantage, building trust, reliability, and differentiation.
Meeting the requirements of ISO 13485 is one thing, putting them into practice is another. That’s why this article is part of a broader series developed for New Zealand’s HealthTech sector, aimed at helping teams turn regulatory expectations into working systems.
This series explores ISO 13485 in four parts: from first steps and system setup to risk management and audit readiness. Participants who complete the quiz at the end of each section will receive an ISO 13485 QMS Certificate of Completion from the HealthTech Activator.
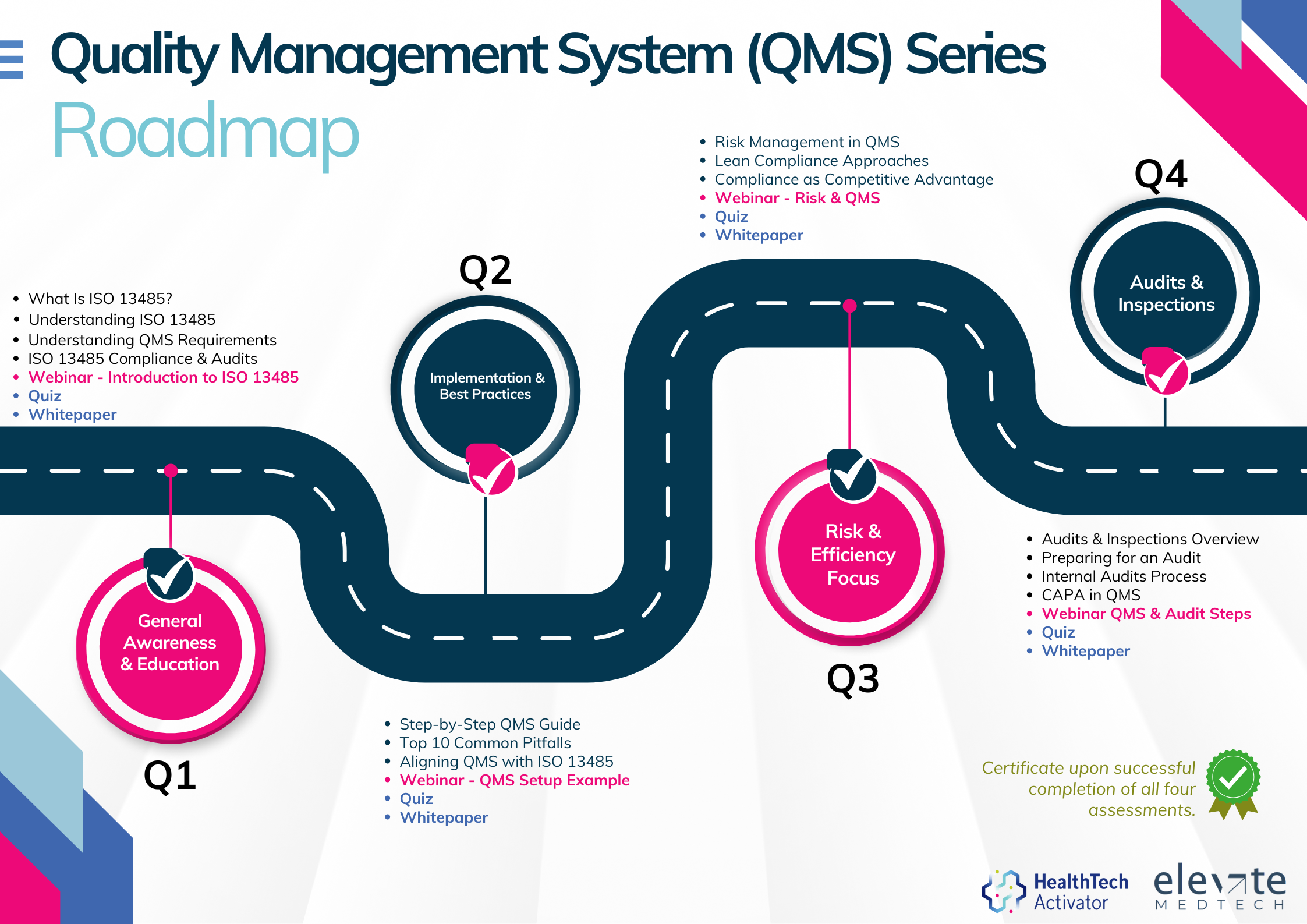
The QMS Series is brought to you by the HealthTech Activator, in partnership with Elevate Medtech.

