Preparing for a Successful ISO 13485 Audit: Checklist and Tips
QMS Series
Regulatory Pathway
Audits
ISO 13485 audits are where your quality management system meets reality. They play a critical role in market access, regulatory compliance, and building trust with customers and partners. More than a formality, they show whether your QMS works in day-to-day operations.
This article ties into the existing quality management systems series and focuses specifically on one question: how can a company prepare for an ISO 13485 audit in a way that makes the audit day structured, confident, and as low‑stress as possible?
The Problem for Companies
For many companies, especially smaller manufacturers or startups, ISO 13485 audits are a major source of stress. Typical problems include:
Documents exist but are hard to find or not up to date.
Processes are followed in practice, but not as they are described in the SOPs.
Roles and responsibilities are unclear – especially who can speak to what in an audit.
Audits are prepared reactively (“tidying up before the visit”) instead of designing the QMS to be audit ready in everyday use.
The result: teams react instead of lead, and unnecessary nonconformities arise, not because the system is broken, but because it cannot be clearly demonstrated.
Background: What the Audit Really Checks
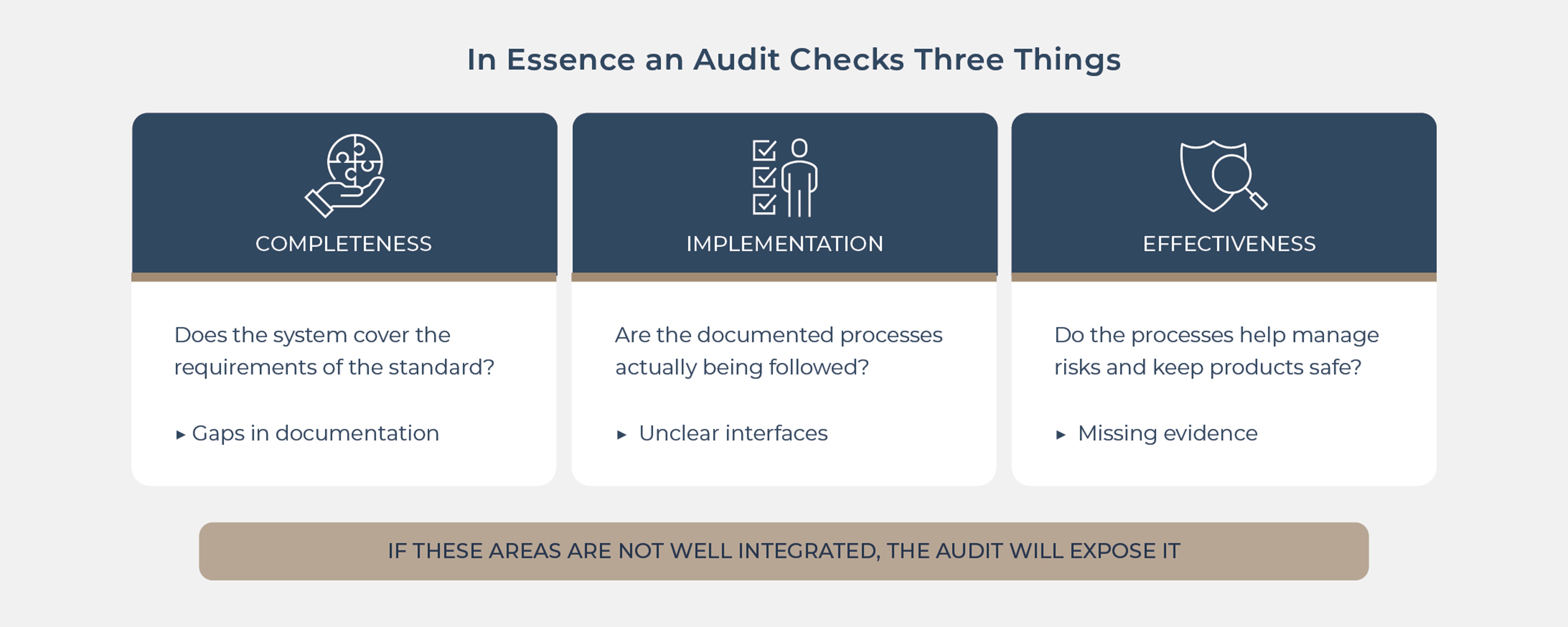
ISO 13485:2016 defines requirements for a QMS that is meant to ensure the safety and performance of medical devices throughout their lifecycle. In essence, an audit checks three things:
Completeness – does your QMS cover the requirements of the standard?
Implementation – are the documented processes actually being followed?
Effectiveness – do the processes manage risk and ensure product safety?
In real-world MedTech, very different functions collide: developmental, clinical, regulatory, quality, and commercial. If these areas are not well integrated, audits will expose gaps in documentation, unclear interfaces, and missing evidence for key decisions.
The good news is that with structured preparation and a pragmatic checklist, ISO 13485 audits can be dramatically dedramatised.
The Solution: Focused, Practical Audit Preparation
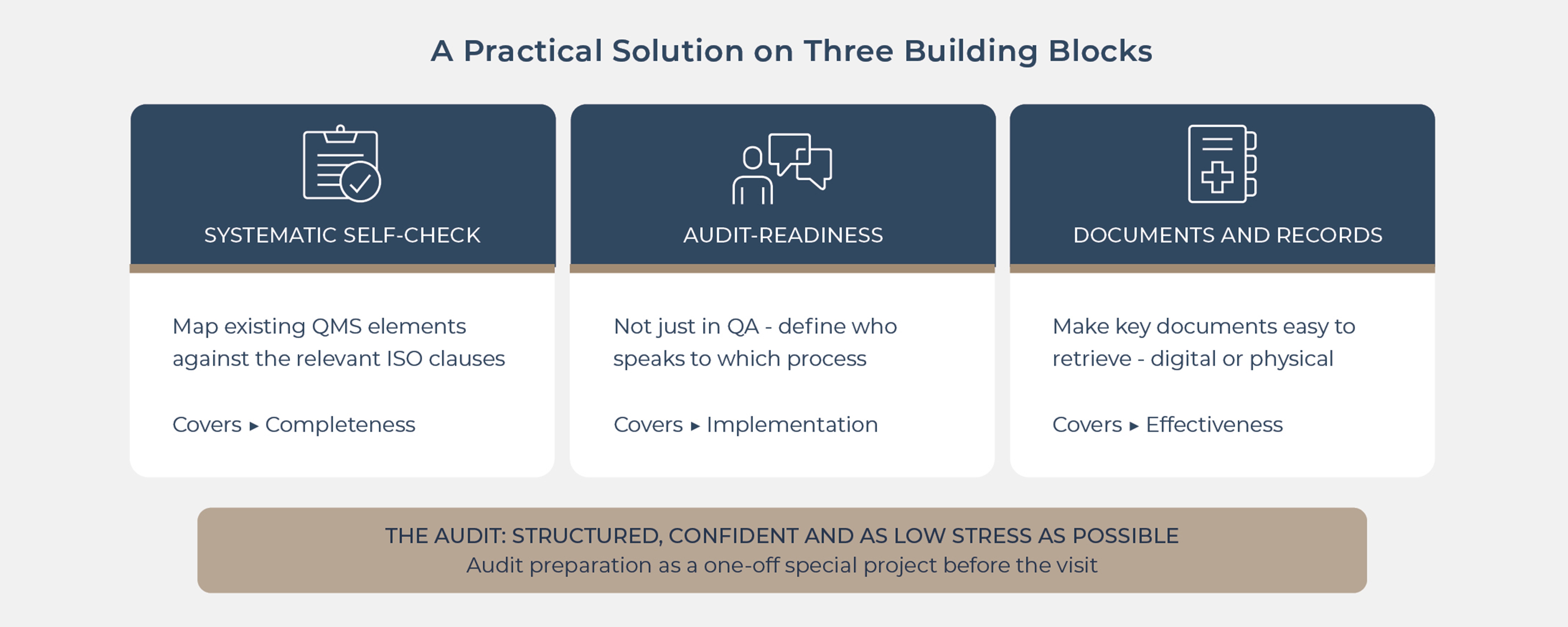
The key is to stop seeing audit preparation as a one-off “special project before the visit” and instead use it to sharpen the focus on what should already be there. A practical solution rests on three building blocks:
Systematic self-check using a checklist
Map existing QMS elements against the relevant ISO 13485 clauses.
Focus on core processes: design and development, document control, risk management, supplier control, production/service, CAPA, and post-market surveillance.
Audit readiness across the team – not just in QA
Define clear roles in the audit. Who speaks to which process?
Hold short, targeted preparation sessions with process owners. What questions are typically asked, and which records will we show?
Make documents and records “audit friendly”
Prepare key documents and records so they are easy to retrieve (digital or physical).
Make the main “storylines” visible: from risk, through requirements and development, to verification/validation, and post-market surveillance.
Practical Application: Checklist and Tips
Below is a practical checklist companies can use to prepare.
1. QMS fundamentals
Is your ISO 13485 certificate status clear (scope, sites, validity)?
Are the quality policy and objectives current, communicated, and understood by staff?
Do you have an up-to-date organisational chart with clear responsibilities, especially for regulatory and quality?
2. Document control
Is there a central, controlled repository for documents (SOPs, forms, work instructions)?
Are versions traceable (revision history, approvals, effective dates)?
Have obsolete documents been removed or clearly marked as “invalid”?
3. Risk management and development
Is there a current risk file for each product, linked to development and post-market surveillance?
Are design and development files complete: requirements, design outputs, verification/validation, design reviews, design transfer?
For software or usability, are deliverables clearly traceable?
4. Supplier and service provider control
Do you maintain a list of qualified suppliers and service providers with risk ratings?
Are supplier evaluations, audits or other monitoring activities documented?
Are quality agreements in place where needed (e.g. with contract manufacturers or critical service providers)?
5. Production, service and traceability
Are manufacturing/service processes described and demonstrably followed (e.g. DHRs, test records)?
Is traceability assured to the required extent (batches, serial numbers, field stock)?
6. CAPA, complaints and PMS
Is there a single system for capturing complaints, nonconformities and field feedback?
Are root cause analyses and CAPA documented and checked for effectiveness?
Are post-market surveillance activities (e.g. regular data reviews, trend analyses) visible, and do they feed back into risk management and development?
7. Internal audits and management review
Do recent internal audit reports cover all relevant QMS areas?
Are management reviews documented with decisions and actions followed through?
8. People and audit communication
Do process owners know that an audit is coming and understand what an auditor will typically want to see?
Are there clear guidelines for audit communication, such as being honest, specific, and evidence-led?
Practical tips for audit day
Show reality, not a performance: Audits are easier when the QMS is genuinely used. Preparation then becomes organising, not firefighting.
Have key evidence ready: For core processes, a simple “audit pack” (digital or physical) with key records can save time and stress.
Stay calm and clarify questions: If a question is unclear, ask. It is better to align than to guess.
By working through these points in advance, companies can shift ISO 13485 audits from a stressful checkpoint into a structured review and use it as a strengthen their quality management system in a meaningful way.
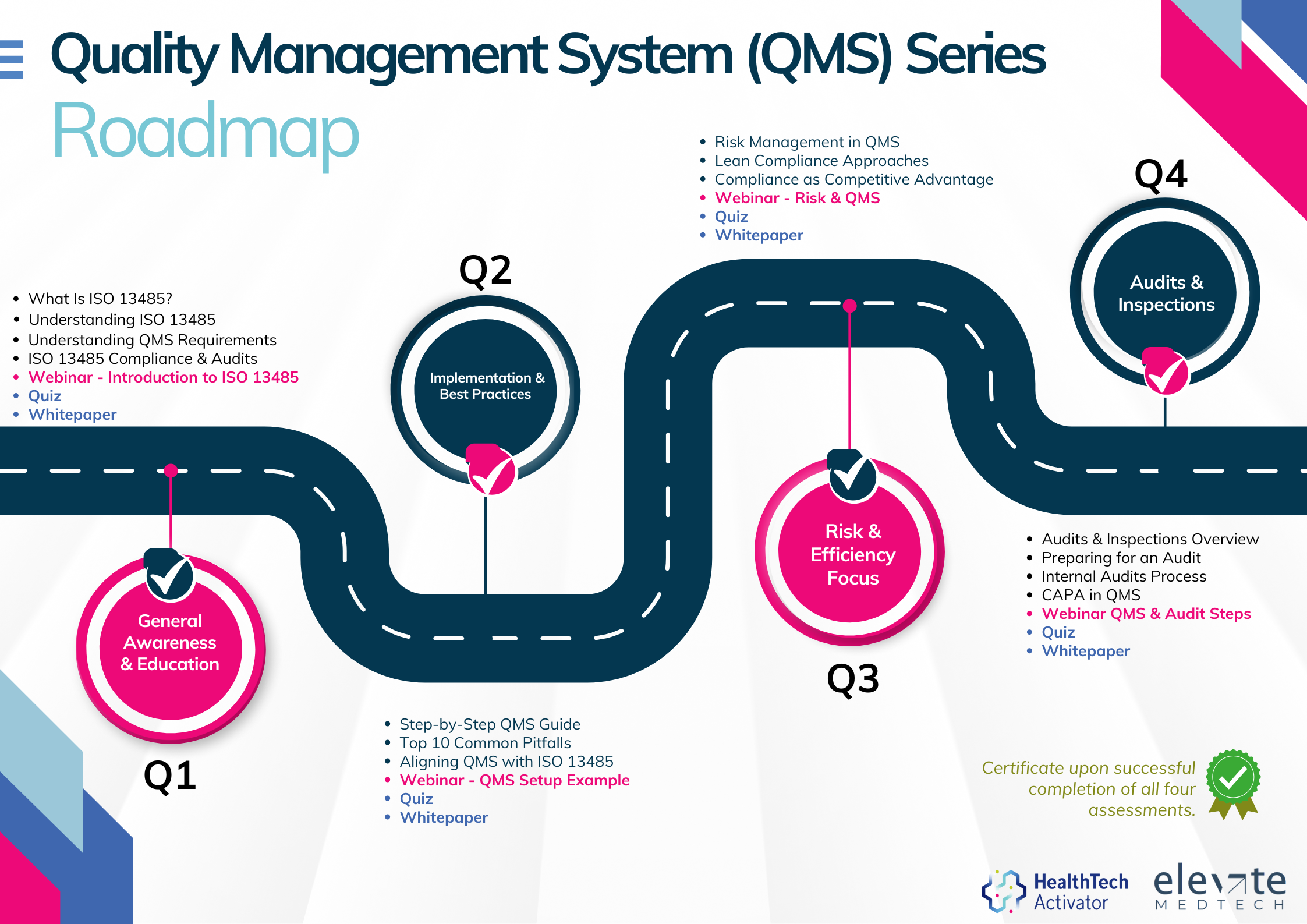
QMS Learning Series
Meeting the requirements of ISO 13485 is one thing, putting them into practice is another. That’s why this article is part of a broader series developed for New Zealand’s HealthTech sector, aimed at helping teams turn regulatory expectations into working systems.
This series explores ISO 13485 in four parts: from first steps and system setup to risk management and audit readiness. Participants who complete the quiz at the end of each section will receive an ISO 13485 QMS Certificate of Completion from the HealthTech Activator.